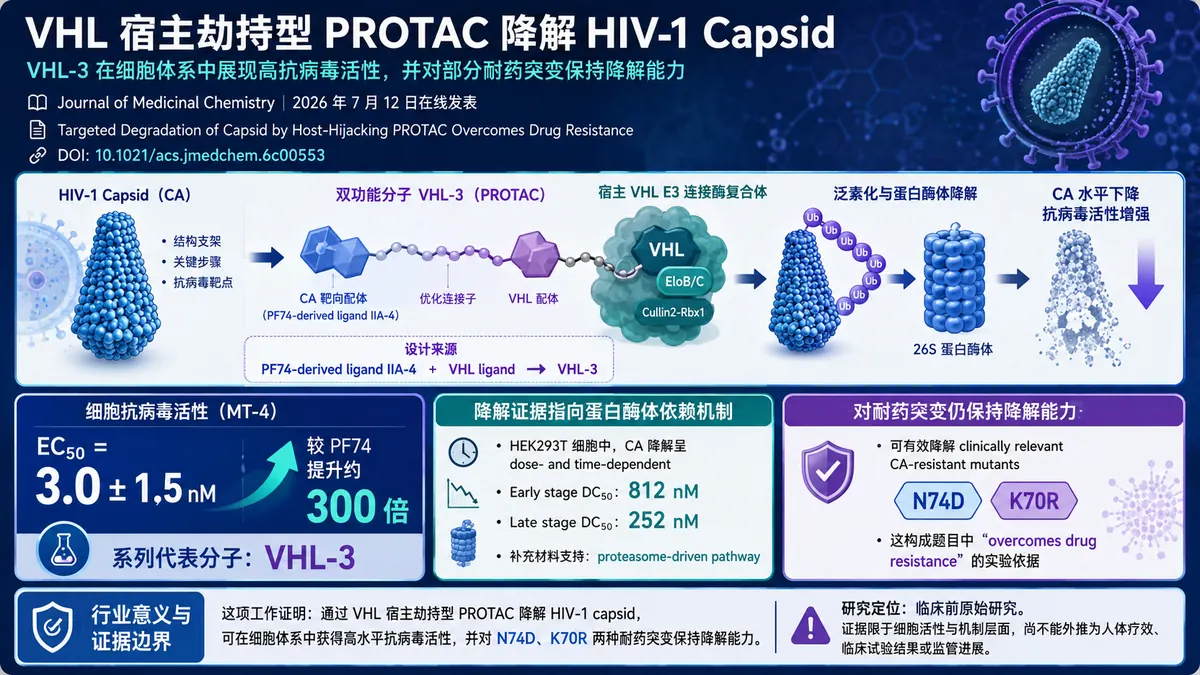
2026 年 7 月 12 日,一篇聚焦 HIV-1 capsid(CA)靶向降解的临床前原始研究在线发表于《Journal of Medicinal Chemistry》。论文题为《Targeted Degradation of Capsid by Host-Hijacking PROTAC Overcomes Drug Resistance》,DOI 为 10.1021/acs.jmedchem.6c00553。研究团队以宿主 E3 连接酶 VHL 为劫持对象,构建了一组面向 HIV-1 CA 的 PROTAC 降解剂,并将 VHL-3 确定为其中具有代表性的活性分子。
从 PF74 衍生配体到 VHL-3
该研究的分子设计以 PF74-derived capsid ligand IIA-4 为靶蛋白结合端,再连接 VHL E3 ligase ligand,形成 CA-targeted PROTAC degraders。其核心思路不是仅依赖配体占据 capsid,而是通过双功能分子同时结合 CA 与 VHL,借助宿主细胞的蛋白降解系统降低 CA 水平。论文标题中的“host-hijacking”即指这种对宿主 VHL E3 连接酶系统的利用。
在该系列化合物中,VHL-3 展现出突出的细胞抗病毒活性。研究给出的结果显示,VHL-3 在 MT-4 细胞中的 anti-HIV-1 活性 EC50 为 3.0 ± 1.5 nM,较 PF74 提升约 300 倍。这一结果表明,将 IIA-4 与 VHL 配体连接后,所得双功能分子在该细胞体系中的活性明显高于作为比较对象的 PF74。
降解证据指向蛋白酶体依赖机制
机制研究在 HEK293T 细胞中展开。结果显示,VHL-3 可随剂量增加和处理时间延长而降低 capsid(CA)水平,即呈现 dose- and time-dependent 的降解特征。研究分别报告了 early stage 与 late stage 条件下的降解效力:early stage DC50 为 812 nM,late stage DC50 为 252 nM。
补充材料进一步明确,VHL-3 引发的 CA 降低属于 proteasome-driven pathway。换言之,研究观察到的效应被归入蛋白酶体驱动的靶蛋白降解,而不是仅以传统抑制剂的占位作用解释。这一点构成该研究最关键的机制证据,也使 VHL-3 与单纯的 capsid 配体区分开来。
值得注意的是,MT-4 细胞中的抗病毒 EC50 与 HEK293T 细胞中的降解 DC50 来自不同实验体系和观察终点,数值不能直接等同或简单换算。现有结果支持“细胞抗病毒活性增强”与“CA 水平下降并由蛋白酶体通路驱动”同时成立,但并不足以单独量化二者之间的因果贡献比例。
对两种 CA 耐药突变仍保持降解能力
耐药相关结果是这篇论文的另一重点。补充材料指出,VHL-3 可有效降解 clinically relevant CA-resistant mutants N74D 与 K70R。对于面向病毒蛋白的降解策略而言,这一结果说明,至少在本研究所采用的细胞和机制实验条件下,两种 CA 耐药突变并未消除 VHL-3 的降解活性。
这也是论文题目中“overcomes drug resistance”的实验依据。不过,这一表述应严格限定在研究已经展示的范围内:VHL-3 对 N74D 和 K70R 突变体仍能实现有效降解,并不等同于已经证明其能够处理所有 capsid 耐药变异,也不能外推为临床患者中的耐药逆转效果。
行业意义与证据边界
- 靶点层面:研究将 HIV-1 capsid 纳入 VHL 招募型 PROTAC 的降解框架,给出了从分子设计、细胞抗病毒活性到靶蛋白下降的连续证据。
- 活性层面:VHL-3 在 MT-4 细胞中的 EC50 达 3.0 ± 1.5 nM,较 PF74 提升约 300 倍,是该系列最受关注的结果之一。
- 机制层面:HEK293T 细胞实验显示 CA 降解具有剂量和时间依赖性,并被明确归为蛋白酶体驱动通路。
- 耐药层面:VHL-3 对 N74D 与 K70R 两种具有临床相关性的 CA 耐药突变体仍保持有效降解。
截至 2026 年 7 月 12 日,这项工作应被定位为 HIV-1 临床前抗病毒原始研究。其证据限于细胞活性与机制层面,尚不能写成人体疗效、临床试验结果或监管进展。VHL-3 是否具备进一步开发价值,仍需更多药代、选择性、安全性、耐药谱及体内研究支持。就当前结果而言,该论文的主要贡献,是证明通过 VHL 宿主劫持型 PROTAC 降解 HIV-1 capsid,在细胞体系中能够获得高水平抗病毒活性,并对 N74D、K70R 两种耐药突变保持降解能力。